New therapeutic candidate for Spinal Muscular Atrophy
Patents for licensing
Development of a therapeutic solution for Spinal Muscular Atrophy (SMA) focused to restore protein SMN levels in neurodegenerative disorder which primarily affecting motor neurons (MNs).
Spinal Muscular Atrophy (SMA) is a genetic neuromuscular disease characterized by muscle atrophy and weakness. The disease generally manifests early in life and is the leading genetic cause of death in infants and toddlers. SMA is caused by defects in the Survival Motor Neuron 1 (SMN1) gene that encodes the SMN protein. The SMN protein is critical to the health and survival of the nerve cells in the spinal cord responsible for muscle contraction (motor neurons).

The Technology
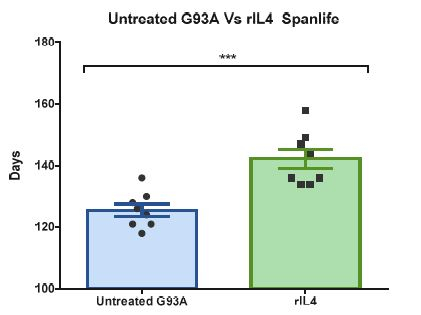
Researchers in the University of Lleida have preclinical evidence for the positive effects of a calpain inhibitor in in vitro and in vivo models for SMA (severe model and mild model for SMA). When treated with this inhibitor, mice survival is substantially prolonged and the clinical course of the disease is improved by means of attenuating body weight loss and motor behaviour (righting reflex and tube test). The treatment increases SMN protein level in motor neurons and also prevents neurite degeneration in SMA mutants MNs.
The Market Need
Disease modifying therapeutic approaches are currently aiming to increase SMN protein levels, using gene therapy to add an exogenous copy of SMN, or protecting motor neurons from degenerating. However, there is no effective treatment for SMA, particularly for the most severe forms.
SMA has generally been believed to affect as many as 10,000 to 25,000 children and adults in the US, and therefore it is one of the most common rare diseases. One in 6,000 to one in 10,000 children are born with the disease. In Spain, there are around 60 new SMA diagnoses every year.
Spinraza© (Nusinersen, Biogen) is the only drug approved by the FDA and EMA, but there are other candidates in the pipeline. The SMA therapeutics pipeline is segmented into early stage candidates and late stage candidates under development, as Spinraza© long-term effects are still unknown.
Advantages
Treatment with a calpain inhibitor in vitro cultures of MNs and in animal models of AME is able to improve survival, the most apparent symptoms and the clinical course of the disease by attenuating weight lossand improving motor behaviour.
Applications
The invention can be used for the treatment of patients who have already shown or who have yet to show symptoms of the disease.
Level of Development
This project is currently in a preclinical stage of development. In vitro and in vivo assays have been performed with this potential therapeutic.
Current development status
Technology available for licensing
The Valorisation and Technology Transfer Unit (UViT) is the specialized unit for handling the IPR portfolio in the University of Lleida. The UViT works close to researchers in the fields of Agrifood, Biomedicine, Technology and Sustainability and Social Development to identify research results with market potential, to assess the community on the best IP protection strategy and to commercialise technologies through licensing-out agreements and the creation of technology-based companies.
Create your free account to connect with Universitat de Lleida and thousands of other innovative organizations and professionals worldwide
Send a request for information
to Universitat de Lleida
Technology Offers on Innoget are directly posted
and managed by its members as well as evaluation of requests for information. Innoget is the trusted open innovation and science network aimed at directly connect industry needs with professionals online.
Need help requesting additional information or have questions regarding this Technology Offer?
Contact Innoget support